

Rice University’s Peter Wolynes and his research team have unveiled a breakthrough in understanding how specific genetic sequences, known as pseudogenes, evolve. Their paper was published May 13 by the Proceedings of the National Academy of Sciences of the United States of America Journal.
Led by Wolynes, the D.R. Bullard-Welch Foundation Professor of Science, professor of chemistry, biosciences and physics and astronomy and co-director of the Center for Theoretical Biological Physics (CTBP), the team focused on deciphering the complex energy landscapes of de-evolved, putative protein sequences corresponding to pseudogenes.

Pseudogenes are segments of DNA that once encoded proteins but have since lost their ability to do so due to sequence degradation — a phenomenon referred to as devolution. Here, devolution represents an unconstrained evolutionary process that occurs without the usual evolutionary pressures that regulate functional protein-coding sequences.
Despite their inactive state, pseudogenes offer a window into the evolutionary journey of proteins.
“Our paper explains that proteins can de-evolve,” Wolynes said. “A DNA sequence can, by mutations or other means, lose the signal that tells it to code for a protein. The DNA continues to mutate but does not have to lead to a sequence that can fold.”
The researchers studied junk DNA in a genome that has de-evolved. Their research revealed that a mutation accumulation in pseudogene sequences typically disrupts the native network of stabilizing interactions, making it challenging for these sequences, if they were to be translated, to fold into functional proteins.
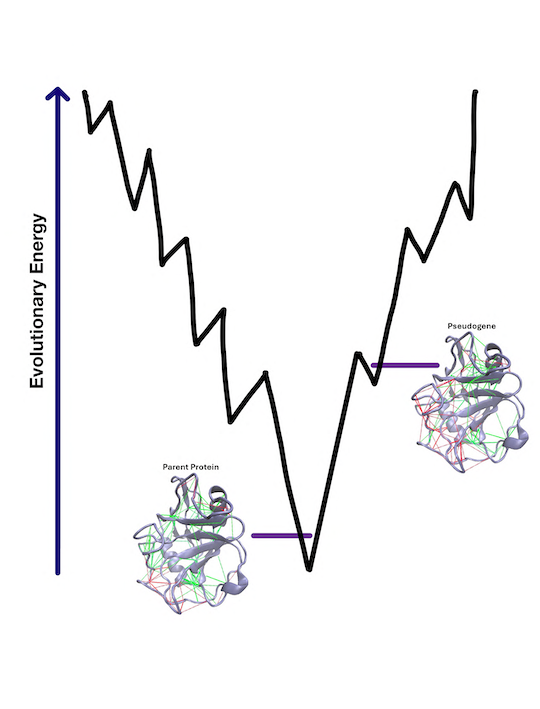
However, the researchers observed instances where certain mutations unexpectedly stabilized the folding of pseudogenes at the cost of altering their previous biological functions.
They identified specific pseudogenes, such as cyclophilin A, profilin-1 and small ubiquitin-like modifier 2 protein, where stabilizing mutations occurred in regions crucial for binding to other molecules and other functions, suggesting a complex balance between protein stability and biological activity.
Moreover, the study highlights the dynamic nature of protein evolution as some previously pseudogenized genes may regain their protein-coding function over time despite undergoing multiple mutations.
Using sophisticated computational models, the researchers interpreted the interplay between physical folding landscapes and the evolutionary landscapes of pseudogenes. Their findings provide evidence that the funnellike character of folding landscapes comes from evolution.
“Proteins can de-evolve and have their ability to fold compromised over time due to mutations or other means,” Wolynes said. “Our study offers the first direct evidence that evolution is shaping the folding of proteins.”
Along with Wolynes, the research team includes lead author and applied physics graduate student Hana Jaafari; CTBP postdoctoral associate Carlos Bueno; University of Texas at Dallas graduate student Jonathan Martin; Faruck Morcos, associate professor in the Department of Biological Sciences at UT-Dallas; and CTBP biophysics researcher Nicholas P. Schafer.
The implications of this research extend beyond theoretical biology with potential applications in protein engineering, Jaafari said.
“It would be interesting to see if someone at a lab could confirm our results to see what happens to the pseudogenes that were more physically stable,” Jaafari said. “We have an idea based on our analysis, but it’d be compelling to get some experimental validation.”