
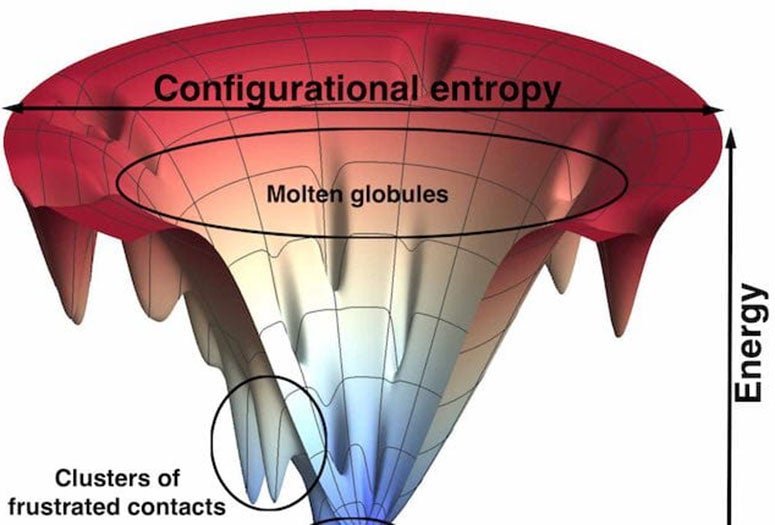
Knowing precisely where proteins are frustrated could go a long way toward making better drugs.
That’s one result of a new study by Rice University scientists looking for the mechanisms that stabilize or destabilize key sections of biomolecules.
Atom-scale models by Rice theorist Peter Wolynes, lead author and alumnus Mingchen Chen and their colleagues at the Center for Theoretical Biological Physics show that not only are some specific frustrated sequences in proteins necessary to allow them to function, locating them also offers clues to achieve better specificity for drugs.
That knowledge could also help design drugs with fewer side effects, Wolynes said.
The team’s open-access study appears in Nature Communications.
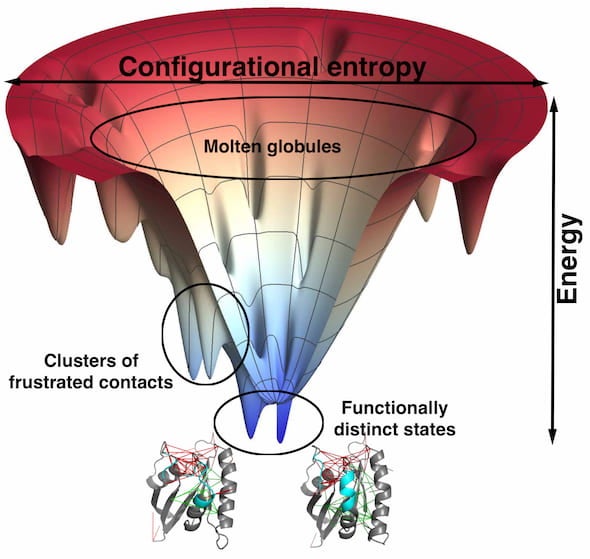
The atom-scale models zero in on the interactions within possible binding sites rather than the vast majority of the interactions in proteins that guide their folding. The finer resolution models allow the incorporation of co-factors like chemically active ligands, including drug molecules. The researchers say this ability gives new insight into why ligands are best captured only by specific proteins and not by others.
“Unnatural ligands,” aka drugs, tend to bind best with those frustrated pockets in proteins that become minimally frustrated once the drugs bind, Wolynes said. Having a way to find and then learn the details of these minimally frustrated sites would help pharmaceutical companies eliminate a lot of trial and error.
“The standard way of doing drug design is to try out 10,000 binding sites on a protein to find ones that fit,” Wolynes said. “We’re saying you don’t have to sample all possible binding sites, just a reasonably fair number to understand the statistics of what could work in local environments.
“It’s the difference between taking a poll and actually having an election,” he said. “The poll is cheaper, but you still will need to check things out.”
The Rice researchers are known for their energy landscape theory of how proteins fold. It usually employs coarse-grained models in which amino acids are represented by just a few sites.
That strategy takes less computing power than trying to determine the positions over time of every atom in every residue, and yet it has proven highly accurate in predicting how proteins fold based on their sequences. But for this study, the researchers modeled proteins and protein-ligand complexes at the atomic level to see if they could find how frustration gives some parts of a protein the flexibility required to bind to other molecules.
“One of the great things about modeling at all-atom resolution is that it allows us to evaluate whether drug molecules fit well into binding sites or not,” Wolynes said. “This method is able to quickly show whether a binding site for a certain drug will be minimally frustrated or will remain a frustrated region. If after the molecule binds the site remains frustrated, the protein could rearrange or the drug could change its orientation in such a way that it could give rise to side effects.”
Modeling the frustrated sites -- and sometimes altering them to see what would happen -- lets the researchers see how drug specificity correlates with binding pockets. Frustration analysis, they wrote, provides “a route for screening for more specific compounds for drug discovery.”
“This concept of frustration was there at the very beginning of our work on protein folding,” Wolynes said. “When we applied it to real protein molecules, we found some examples where the mechanism of folding violated what we would predict from a perfect funnel. Then we discovered these deviations from the funnel picture occurred where the protein was, in fact, somewhat frustrated.
“It was like the exception that proves the rule,” he said. “Something that’s true all the time might be trivial. But if it’s not true 1% of the time, it’s a problem to be solved, and we’ve been able to do that with AWSEM, our structure-prediction software.”
Extending the software to analyze frustration on the atomic level is possible, as described by the group in another recent paper. But the computational cost of tracking every atom in a protein is so high that the researchers needed a way to sample the motions of specific regions where frustration might confuse the folding route.
“Mingchen realized there was an efficient algorithm to sample the local environments in binding sites but keep the atomistic resolution,” said Wolynes, who noted he and Chen, now in private industry, are using the models to investigate possible therapeutics, including COVID-19-related drugs.
Co-authors of the paper are Rice graduate student Xun Chen, alumnus Nicholas Schafer and Cecilia Clementi, a former Rice professor and now the Einstein Professor of Physics at the Free University of Berlin; Elizabeth Komives, a professor of chemistry and biochemistry at the University of California, San Diego; and Diego Ferreiro, a biological chemist at the University of Buenos Aires. Wolynes is the D.R. Bullard-Welch Foundation Professor of Science, Professor of Chemistry, BioSciences, and Physics and Astronomy at Rice and co-director of the CTBP.
The National Science Foundation supported the research.