SUMMARY: Rice University chemists find new clues to why proteins misfold. Their theoretical work has implications for degenerative diseases associated with the aggregation of amyloid fibers, like Alzheimer’s and Parkinson’s.
Mike Williams
713-348-6728
mikewilliams@rice.edu
Rice researchers see surprising twist to protein misfolding
AWSEM software finds new clues to degenerative diseases
HOUSTON – (Jan. 14, 2013) – An effort to develop software that unravels the complexities of how proteins fold is paying dividends in new findings on how they misfold, according to researchers at Rice University.
The study published this week in the Proceedings of the National Academy of Sciences by chemist Peter Wolynes and his team at Rice’s BioScience Research Collaborative should be of particular interest to those who probe the roots of degenerative diseases associated with the aggregation of amyloid fibers in the body. These include Alzheimer’s and Parkinson’s diseases and Type 2 diabetes.
The molecular dynamics software to predict how strands of residues bend and twist into their functional shapes is designed to follow Wolynes’ and his colleagues’ groundbreaking “principle of minimal frustration.” These residues, the molecular beads that make up proteins, follow the path of least resistance as they fold into their native states. The principle describes how evolution has shaped the path a protein takes toward stability.
The software, called AWSEM-MD (for associative memory, water-mediated structure and energy model) simulates the possible ways beads in a strand should fold, based on the energies at play down to the submolecular level, and accurately predicts the final structure. Two developers of the current version, Weihua Zheng, a postdoctoral researcher at Rice, and Nicholas Schafer, a graduate student, are co-authors of the new paper, the latest in a series on folding dynamics dependent on the software.
The researchers set out to confirm that a process seen by experimentalists called domain swapping is one cause of protein misfolding. Domains are conserved parts of protein chains. Occasionally, a domain in one chain may encounter its doppelgänger in a nearby chain and become entangled with it via interactions similar to those in the correctly folded state.
The result is often a dimer – a kind of protein Siamese twin – that probably won’t be able to perform its intended biological task and may become part of a damaging amyloid fibril. “Experimentalists had some strong laboratory evidence that dimerization is a consequence of minimal frustration, an idea proposed earlier by our group on more general grounds,” Wolynes said. “So we figured it would be nice to do a simulation to check it.”
The team did indeed see domain swapping in their models of human cardiac titin, a muscle protein. But they were surprised to see something they weren’t looking for: evidence that identical sequences in neighboring chains, as short as five to seven residues, had the unfortunate tendency to find each other and stick together.
They found instances of such “self-recognition” tipped the balance of energies that dictated whether a protein would fold properly. Replacing just a few residues in one fragment eliminated self-recognition and lowered the incidence of domain swapping, Wolynes said.
“We weren’t the people who thought of this as a possibility,” he said. “It had been suggested by others, although I never really believed it because it doesn’t have an obvious connection to the principle of minimal frustration.” But the simulations showed instances where sticky self-recognition in one segment of a chain could affect the energy of residues down the line and effectively introduce “frustration” that keeps the rest of the protein from folding at all and results in high disorder, or entropy.
While the models don’t directly connect to the formation of amyloid fibrils, Wolynes said, anecdotal evidence indicates protein-folding diseases have some correlation with fevers that allow the extra entropy to stabilize the misfolded forms. “Our results would provide a new explanation,” he said, for how a disordered part of the chain can contribute to the stability of these misfolded states at high temperature.
“When you hear ‘take two aspirins and call me in the morning,’ your doctor is doing you a bigger favor than you know,” Wolynes said.
The discovery could open paths for researchers to design drugs that inhibit specific interactions. “Very minor changes seem to destroy this self-recognition in the computer simulation, and that’s what we want the experimentalists to do: Make those changes to see if they decrease the self-recognition effect,” he said.
“Our simulations provide structural details of misfolded proteins at the molecular level that are difficult for experiments to probe,” Zheng said. “These can generate specific hypotheses they can test.”
The researchers hope their work will be useful to both experimentalists and other computational protein-folding researchers.
“AWSEM is hosted on Google Code, which requires all code to be open-source,” Schafer said. “So it’s available to anyone who wants to use it. What we’re seeing with these studies is that the values we get by applying the principle of minimal frustration are appropriate globally, not just for predicting the native structures of proteins. It can predict bound structures (like dimers) and misfolded structures as well.
“You always have to be careful about using models that ‘rig the deck’ in favor of a particular anticipated result,” he said. “But what’s interesting is that our model doesn’t have any information a priori about these specific types of misfolded structures. Our model is parameterized using as input only experimental data for properly folded structures and then applying the principle of minimal frustration. The wide range of successes we’ve had this year tells me that we have a decent method for deriving the strengths of the interactions.”
“We never really thought about specific kinds of misfolding or the aggregation process when we built our model around the principle of minimal frustration,” Zheng said. “But they all fall into place.”
Garegin Papoian, the Monroe Martin Associate Professor at the University of Maryland, and Aram Davtyan, a graduate student in his lab, designed and programmed most of the current version of AWSEM; this version built upon previous development by Cecilia Clementi, the Wiess Career Development Chair and a professor of chemistry and of chemical and biomolecular engineering at Rice, and others in Wolynes’ labs at the University of California at San Diego and the University of Illinois at Urbana-Champaign.
The National Institute of General Medical Sciences, one of the National Institutes of Health, and the D.R. Bullard-Welch Chair at Rice University supported the research. Wolynes is the Bullard-Welch Foundation Professor of Science and a professor of chemistry and a senior scientist with the Center for Theoretical Biological Physics at Rice. The researchers utilized the SUG@R (Shared University Grid at Rice) and DAVinCI (Data Analysis and Visualization Cyberinfrastructure) at Rice’s Ken Kennedy Institute for Information Technology to run their models.
-30-
Read the paper upon publication at www.pnas.org/cgi/doi/10.1073/pnas.1222130110.
An American Chemical Society video about Wolynes’ protein theory appears here:
http://www.youtube.com/watch?v=N6Al_kqmFDw.
This news release can be found online at news-network.rice.edu/news.
Follow Rice News and Media Relations via Twitter @RiceUNews.
Related Materials:
AWSEM-MD downloads: http://code.google.com/p/awsemmd/.
AWSEM-MD: Protein Structure Prediction Using Coarse-Grained Physical Potentials and Bioinformatically Based Local Structure Biasing:
http://pubs.acs.org/doi/abs/10.1021/jp212541y.
Predictive energy landscapes for protein–protein association:
http://www.pnas.org/content/109/47/19244.
Funnels, Pathways, and the Energy Landscape of Protein Folding: A Synthesis: http://onlinelibrary.wiley.com/doi/10.1002/prot.340210302/abstract;jsessionid=9090D4EA3FFA5424AEEB810614E523C3.d01t04.
Wolynes Research Lab: http://wolynes.rice.edu/node/2?destination=node/2.
Center for Theoretical Biological Physics: http://ctbp.rice.edu/.
Images for download:

https://news2.rice.edu/files/2013/01/PROTEIN-2-WEB.jpg
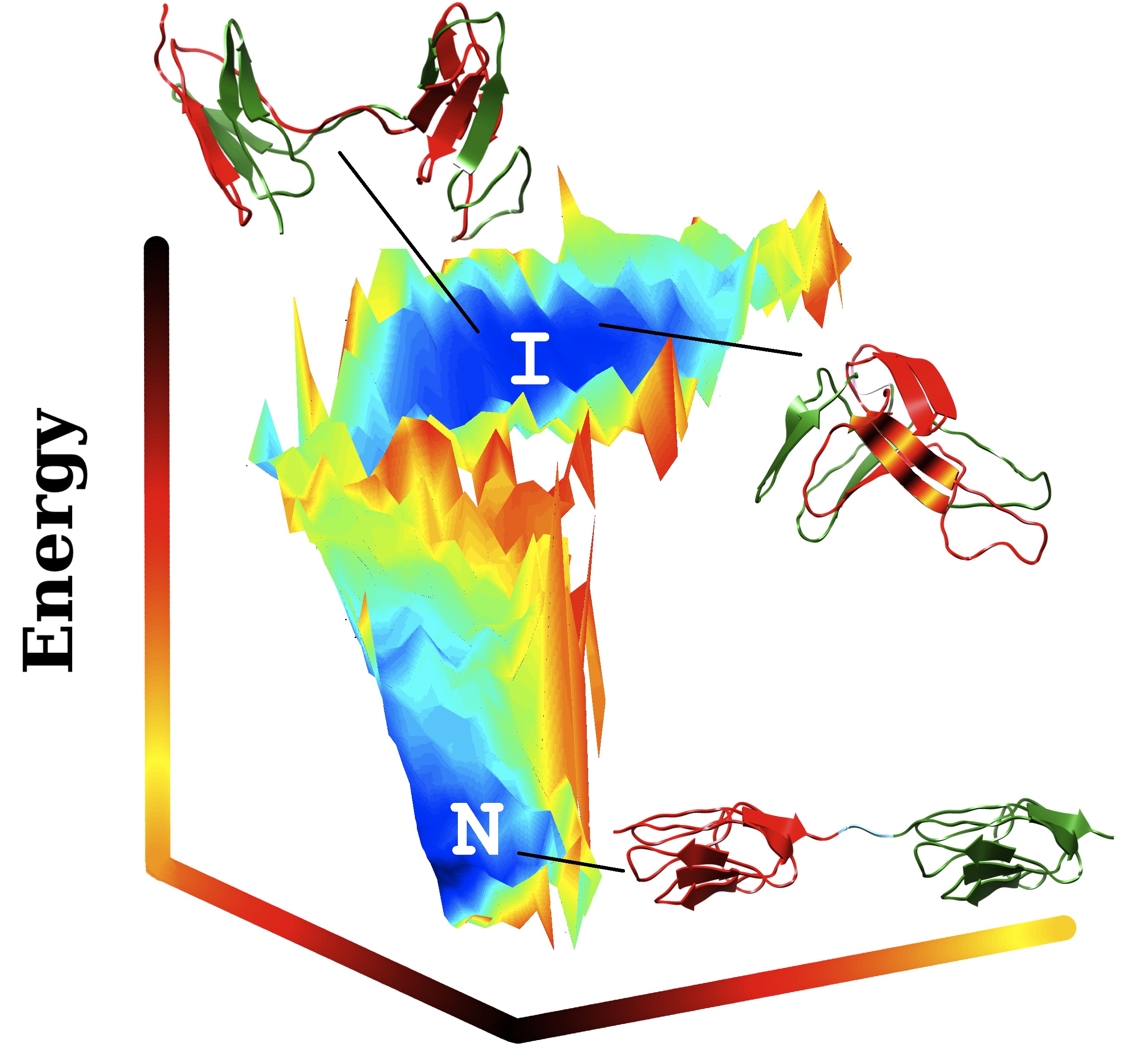
A three-dimensional plot of energies in multidomain protein folding, produced at Rice University with the AWSEM-MD program, shows stable regions in blue at the bottom (“N”) for the protein in its correct native state, and at the top (“I”) in its incorrect, misfolded state. Both extremes are equally stable, though “N” proteins are lower in energy. Red and green represent the separate protein domains; the colors are jumbled in the domain-swapped structure (top) and in the “self-recognizing” structure (right), which have misfolded due to strong self-recognition interactions among the short residue sequences shown in the colored stripes. Researchers suspect misfolded proteins play a major role in the aggregation of amyloid fibers implicated in degenerative diseases. (Credit: Nicholas Schafer/Weihua Zheng/Rice University)

https://news2.rice.edu/files/2013/01/PROTEIN-1-WEB.jpg
Rice researchers, from left, Weihua Zheng, Professor Peter Wolynes and Nicholas Schafer confirmed through their computer models that “self-recognition” among short residue sequences on neighboring proteins plays a role in misfolding that may lead to aggregation. The AWSEM molecular dynamics software developed over many years by Wolynes’ lab simulates the possible ways proteins may fold based on submolecular energies. (Credit: Eva Tittel/Rice University)
Located on a 300-acre forested campus in Houston, Rice University is consistently ranked among the nation’s top 20 universities by U.S. News & World Report. Rice has highly respected schools of Architecture, Business, Continuing Studies, Engineering, Humanities, Music, Natural Sciences and Social Sciences and is home to the Baker Institute for Public Policy. With 3,708 undergraduates and 2,374 graduate students, Rice’s undergraduate student-to-faculty ratio is 6-to-1. Its residential college system builds close-knit communities and lifelong friendships, just one reason why Rice has been ranked No. 1 for best quality of life multiple times by the Princeton Review and No. 2 for “best value” among private universities by Kiplinger’s Personal Finance. To read “What they’re saying about Rice,” go to http://tinyurl.com/AboutRice.
If you do not wish to receive news releases from Rice University, reply to this email and write “unsubscribe” in the subject line. Office of News and Media Relations – MS 300, Rice University, 6100 Main St., Houston.


{kind=link}
{kind=link}