Genomes of limpet, leech and worm put spotlight on lophotrochozoans
A new report in the journal Nature unveils three of the first genomes from a vast, understudied swath of the animal kingdom that includes as many as one-quarter of Earth’s marine species. By publishing the genomes of a leech, an ocean-dwelling worm and a kind of sea snail creature called a limpet, scientists from Rice University, the University of California-Berkeley and the Department of Energy’s Joint Genome Institute (JGI) have more than doubled the number of genomes from a diverse group of animals called lophotrochozoans (pronounced: LOH-foh-troh-coh-zoh-uhns).

Owl limpet
Lophotrochozoans are a diverse group of animals that includes mollusks – such as snails, clams and octopuses — and annelids — such as leeches and earthworms. Like humans and all other animals, lophotrochozoans can trace their evolutionary history to the earliest multicellular creatures. But the lophotrochozoan branch of the evolutionary tree diverged more than 500 million years ago from the branch that produced humans.
“Most animals, including people, have body plans with bilateral symmetry, which means they have left and right sides that are mirror images of one other,” said co-author Nicholas Putnam, assistant professor of ecology and evolutionary biology at Rice. “When you look at all bilaterian species, you can divide them into three big groups that biologists call clades.
“Lophotrochozoans are one of these clades, and when we looked at all of the genomes that had been sequenced, we found that only two were lophotrochozoans,” he said. “That left a big hole in the genetic record, and our goal with this study was to fill in some of the gaps in that blank space.”

Nicholas Putnam
Genome sequencing for the new study was performed at JGI in Walnut Creek, Calif. The three newly sequenced species are Capitella teleta, an ocean-dwelling worm; Helobdella robusta, a freshwater leech; and Lottia gigantean, a large marine mollusk.
Putnam’s co-investigators included Daniel Rokhsar, of both Berkeley and JGI; David Weisblat and Eric Edsinger-Gonzales, both of Berkeley; Elaine Seaver of the University of Hawaii; and Oleg Simakov of the European Molecular Biology Laboratory at Heidelberg, Germany. Putnam’s group, which included Rice graduate student Jie Lv and scientific programmer Paul Havlak, focused on genomic analysis.
“At Rice, we work on comparative genomics,” Putnam said. “We look for recognizable similarities across genomes, and we are interested in similarities among the genes themselves and also among the patterns of genetic organization. These structural similarities can tell us a lot about the evolution of individual genes and functional gene groups, like chromosomes.”
Almost all published genomes are from the animal kingdom’s most-studied clades: deuterostomes, which includes humans and other vertebrates, and ecdysozoans, which includes insects. Only two lophotrochozoan genomes have been previously mapped, and both are for parasitic worms, which aren’t representative of most species in the clade, Putnam said.

The freshwater leech Helobdella robusta
In examining the lophotrochozoan genomes, Putnam’s group developed new computational tools to examine and compare the differences among the three new genomes and hundreds of known genomes, including those for humans and oft-studied model organisms like the fruit fly.
The tools streamlined the search for similarities among the genomes and also helped the researchers trace evolutionary changes that occurred in specific groups of genes.
“Using these tools, we focus on the part of the evolutionary tree where specific mutations took place,” Putnam said. “For example, we can say, ‘A mutation occurred here that moved a big chunk of this chromosome, and it must have happened after lophotrochozoans diverged from deuterostomes but before the split between mollusks and annelids.'”
So far, Putnam’s group has traced 17 “ancestral linkage groups,” large groups of genes that are similar in structure to chromosomes, to the last common ancestor of all the bilaterians.
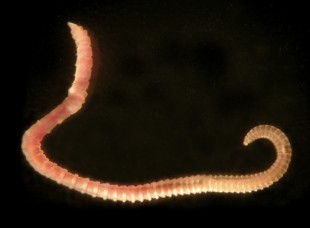
The ocean-dwelling worm Capitella teleta
“For us, the interesting thing is finding genes that exist today in different species, tracing those to a single gene in a common ancestor and then using the patterns we find to test hypotheses about evolutionary processes,” Putnam said. “Sometimes the genes today might still have the same function, but other times they have evolved an entirely new function.”
Putnam said the team has been able to trace the lineage of almost half of the genes in the three new genomes, and the work continues.
“These studies teach us important lessons about ourselves and other vertebrates,” Putnam said. “Looking across the animal kingdom gives us more resolution and clarity about what in our genome is new and what has been preserved from our ancient ancestors.”
Additional co-authors include Ferdinand Marletaz, Tomas Larsson and Detlev Arendt, all of the European Molecular Biology Laboratory; Sung-Jin Cho, Dian-Han Kuo and David Lindberg, all of UC-Berkeley; Uffe Hellsten, Jarrod Chapman, Harris Shapiro, Andrea Aerts, Robert Otillar, Astrid Terry, Jeffrey Boore and Igor Grigoriev, all of JGI; Robert Savage of Williams College at Williamstown, Mass.; Kazutoyo Osoegawa and Pieter de Jong, both of Children’s Hospital Oakland Research Institute in Oakland, Calif.; and Jane Grimwood of both JGI and Hudson Alpha Institute for Biotechnology in Huntsville, Ala.
The research was supported by DOE, the Gordon and Betty Moore Foundation, R. Melmon, the National Science Foundation, the National Institutes of Health, the Boehringer Ingelheim Fonds and the National Human Genome Research Institute.


Thank you so much …. I really appreciate what you do
Thanks for sharing.. its really useful…